
Research History In 2007, we first reported the discovery of a deletion encompassing the first exon of PTCHD1 in a boy with autism as part of a larger study using genotyping microarrays to identify copy number variants in 429 individuals with autism spectrum disorder at the XVth World Congress on Psychiatric Genetics in New York (Noor et al, 2007; noor 2007 wcpg), and then subsequently published under peer review in the American Journal of Human Genetics (Marshall et al, 2008: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2426913/). Dr. Stephen Scherer’s group at the Hospital for Sick Children had generated the microarray data for the autism cohort. Following a long-standing interest of mine in the X-chromosome and autism since working with Profs Hugh Gurling, Patrick Bolton and Mike Rutter back in the early 1990s, we arranged for Abdul Noor, who was my Ph.D. student from 2006-2012, to use the microarray data to identify copy number variants (CNV) of interest on the X-chromosome, as part of his thesis work. This work was supported by a grant from Autism Speaks and a Young Investigator Award from the National Alliance for Research on Schizophrenia and Depression (awarded to Dr. Vincent). Abdul, now Dr. Noor, completed his Ph.D. in 2012, and his thesis is publicly available at https://tspace.library.utoronto.ca/handle/1807/32783.
Among the CNVs of interest identified in this study were a duplication at the gene TSPAN7 (Noor et al, 2009; PMID:19339915), and deletions at IL1RAPL1 and PTCHD1. These findings were included in the Marshall et al 2008 paper, which included findings from all the autosomes as well as our results from the X chromosome. Since mutations at IL1RAPL1 had already been reported for autism and intellectual disability (ID), and the duplication identified at TSPAN7 did not appear to affect the TSPAN7 mRNA (Noor et al, 2007), as a follow up to the Marshall et al study and for Abdul’s thesis work, we decided to focus just on PTCHD1. We subsequently identified several additional autism or ID families with PTCHD1 exonic deletions, as well as autism or ID families with missense variants within PTCHD1. Microarray data from the large Autism Genome Project (AGP) consortium study was available for analysis for this study, in addition to several control cohort data sets, and we reported many deletions in a region just upstream of PTCHD1 that encompassed exons of a long non-coding RNA (PTCHD1AS1-3), putative regulatory elements and in some cases the gene DDX53. While the exonic deletions seemed to be inherited in an X-linked (recessive) Mendelian fashion and segregating fully with the phenotype, the upstream deletions and missense variants showed more complex patterns of inheritance. We also reported the first molecular studies of the PTCHD1 protein, which showed it to be a membrane protein, and showed evidence consistent with a role for PTCHD1 protein as a receptor in the Hedgehog signaling pathway, using transfection of a PTCHD1 construct into Hedgehog-responsive cells, in the presence of Smoothened agonist purmorphamine, and measuring repression of Gli transcription (Noor et al, 2010: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2987731/). This seems to suggest a role for PTCHD1 in Hedgehog signaling, although it cannot be determined at this point whether this is its main function. 
Around the same time, the AGP also published its whole genome analysis of around 1000 autism patients, including the observation that copy number variation in and around the PTCHD1/DDX53 locus showed amongst the most significant findings for autism (Pinto et al, 2010: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3021798/). Also, the iGOLD (Genetics of Learning Disorder) consortium study published their results of a large-scale survey of CNV in 251 intellectual disability families, also including one of the families also described in Noor et al, 2010 (Whibley et al, 2010: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2917707/). icles/PMC3021798/).
Clinical study of PTCHD1 deletion CNVs and truncating mutations
We have recently published our study on the relationship between deletions or truncating mutations at PTCHD1 and clinical features – the first such paper of its kind for PTCHD1- in the journal Clinical Genetics (Chaudhry et al [epub ahead of print]). This paper is available online, although it’s not currently open access: http://www.ncbi.nlm.nih.gov/pubmed/25131214; http://onlinelibrary.wiley.com/doi/10.1111/cge.12482/abstract. In this study, through a large collaborative network of researchers, we have identified 17 families with a PTCHD1 deletion, and report on the clinical features for 13 of these families. We also identified three families with truncating mutations within the PTCHD1 coding region. In these families there is either a single base-pair deletion or single base-pair insertion that disrupts the coding reading frame and leads to a premature STOP codon, thus prematurely truncating the translation from mRNA into protein. In addition to resulting in a shortened protein, such mutations typically trigger a mechanism known as nonsense-mediated mRNA decay (NMD), which targets the aberrant mRNA containing the premature STOP codon for degradation. This mechanism would deplete the PTCHD1 mRNA available for translation to protein. In total, we studied 22 males and 1 female with PTCHD1 mutations. 40% of the subjects had autism spectrum disorder (ASD) or ASD-like behaviours. Global developmental delay was reported for 18 of the 23 (78%). Orofacial hypotonia was noted in 11 of the 23. Generalized hypotonia was found in six, and mild peripheral hypotonia in a further two. There were very few significant medical co-morbidities. Eight reported mild problems with vision, including strabismus (three), jerky oculomotor movements (one), cataracts (one), astigmatism and myopia (one). One individual had mild hearing loss due to recurrent ear infections. Kyphosis was present in one family, mild scoliosis in another. Pes planus with small joint hypermobility was present in one case. One individual had a diagnosis of celiac disease, and one had vesico-ureteral refulux and unilateral inguinal hernia, requiring surgical repair. There was no easily distinguishable facial gestalt that would point to a PTCHD1 mutation diagnosis, however, subtle facial features were noted in some, in particular relating to the mid-facial hypotonia. Lip and mouth shape may also be relevant, but only subtly, and not in every case. We hope that our study paves the way for a more detailed study, incorporating more PTCHD1 patients, and featuring a more detailed clinical and physical analysis. We also hope to conduct a similar analysis for subjects with either deletions just upstream of PTCHD1, as reported in Noor et al, 2010, also for subjects with missense variants within PTCHD1. However, for the latter, we need to find a method for assessing PTCHD1 function (and indeed some clear functionality!) so that the effects of missense changes may be evaluated empirically. This paper was presented as a poster at the World Congress on Psychiatric Genetics, in Copenhagen, Oct 2014, and at the American Society for Human Genetics, San Diego, October 2014. The poster is attached below: PTCHD1 poster
PTCHD1 is a member of the Patched-domain containing family of proteins
The PTCHD1 protein shows homology with the known Hedgehog-signalling receptors PTCH1 and PTCH2. In addition, there is also homology with the Niemann-Pick type C protein 1, as well as PTCHD2, 3 and 4.

These proteins contain a sterol sensing domain. In the case of NPC1, this domain is involved in trafficking intracellular cholesterol, and mutations in NPC1 lead to a lysosomal storage disease. PTCH1 and PTCH2 both bind Sonic Hedgehog, with the two large external loops forming the ligand binding site. The PTCHD1 (large isoform, 888 amino acids) also contains two external loops. The sterol –sensing domain in PTCHD1 spans a number of the predicted transmembrane domains. Several cholesterol binding motifs and an oxysterol binding motif, as predicted using the MiniMotif Miner algorithm. A proposed model of the PTCHD1 protein is shown below.
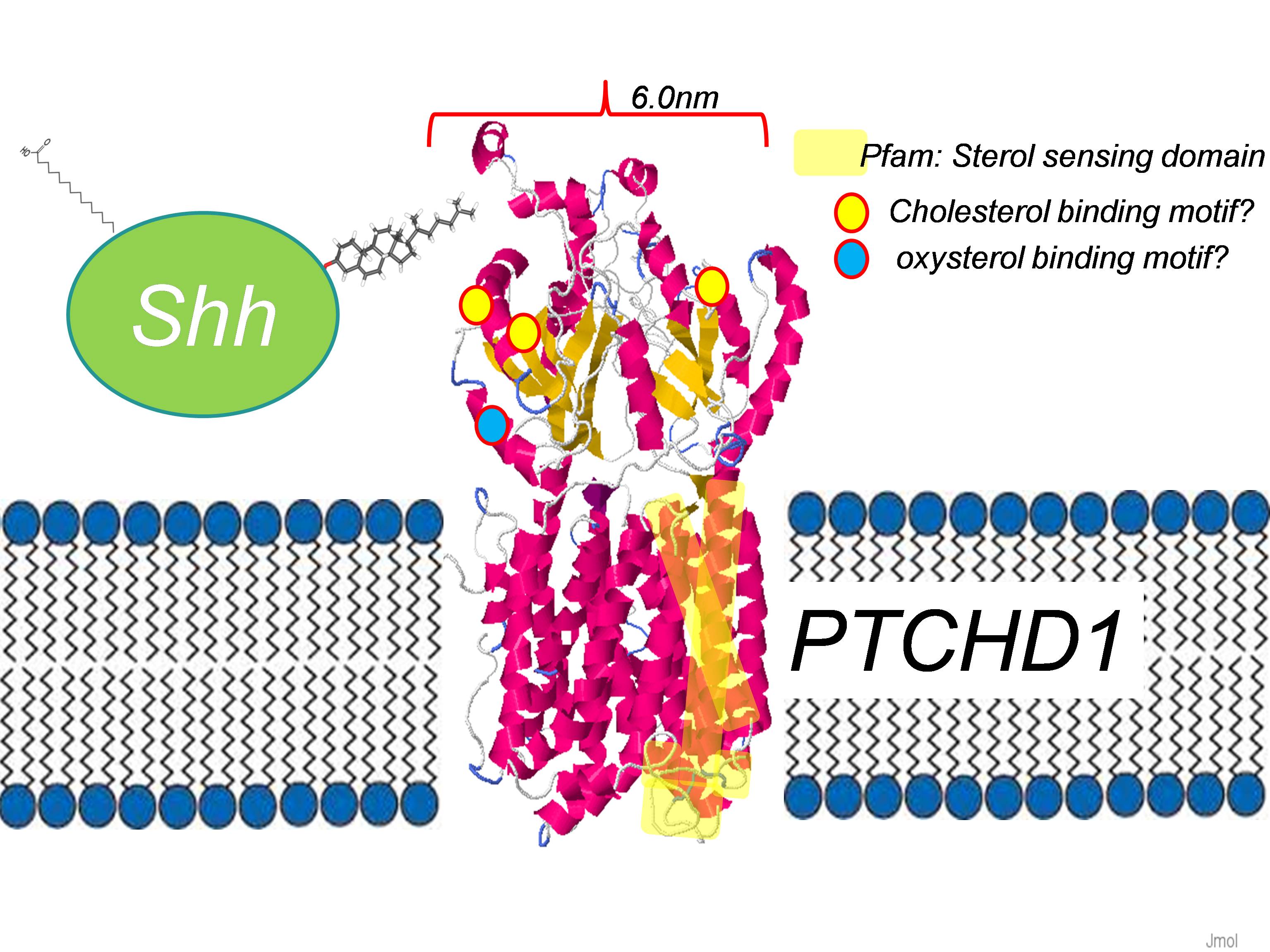
Evolutionary comparison of PTCHD1 orthologues (updated 6 March 2015)
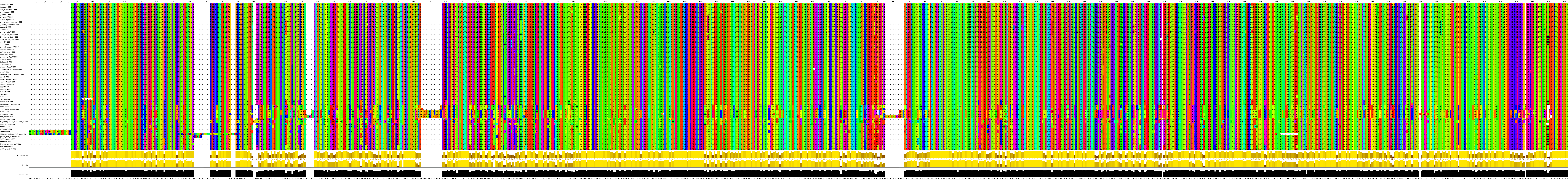
We compared predicted PTCHD1 orthologues from species spanning vertebrate evolution, using CLUSTALW2 sequence alignment. This shows strong conservation throughout vertebrate evolution. Interestingly, no orthologues were identified in any non-vertebrate species, nor in the archao-chordate (amphioxiform order), Branchiostoma floridae (lancelet). The alignment below is displayed using JALVIEW, with “Taylor” amino acid designations (click and zoom below to view).
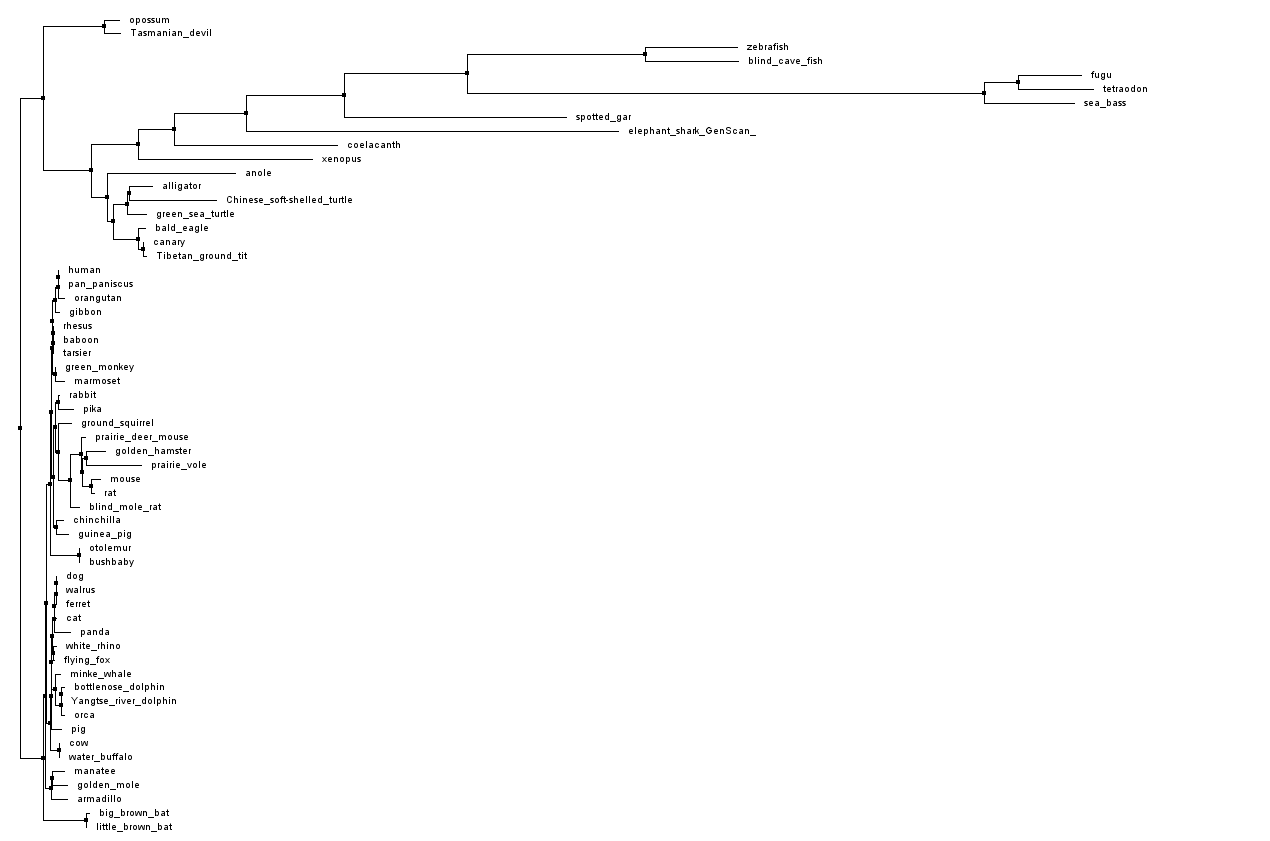
A phylogenetic tree showing the relationships between PTCHD1 orthologues was calculated through Jalview using % identity, by Neighbour Joining method, secondly by Average Distance:
The Ka/Ks ratio for a given gene is the ratio of the number of nonsynonymous nucleotide substitutions per nonsynonymous site (Ka) to the number of synonymous substitutions per synonymous site (Ks). Calculation of Ka/Ks ratios can be used to indicate what type of selective pressure has acted on the gene across evolution, or during a specific period of evolution. Analysis of the nucleotide sequences used for PTCHD1 orthologues from the species listed above suggests that PTCHD1 is under stabilizing (or purifying) selection. This is the most common type of natural selection, and suggest that extremes of traits due to PTCHD1 expression, as the result of changes in coding sequence, undergo negative selection:
tau_ptchd1 (from Selection server, Stern et al, 2007; http://selecton.tau.ac.il)
K+/activity-dependent activation of PTCHD1 expression
A study published in Nature in 2010 from Michael Greenberg’s group looked at genes for which activity in neuronal cultures was dependent on K+ stimulation (Kim et al, 2010: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3020079/). The data shows that PTCHD1 is transcriptionally upregulated (3-fold) upon culturing primary neurons in KCl for 6hrs. This could indicate that PTCHD1 function is dependent on neuronal activity, perhaps during memory and learning-related activities.
Co-expression studies in mouse brain
A recent study by Menashe and colleagues looked at the co-expression network of 26 autism genes from AutDB (http://mindspec.org/autdb.html), including Ptchd1, using data from mouse in situ hybridization studies, suggesting a high degree of correlation of expression of Ptchd1 with other autism genes, and that Ptchd1 sits in a statistically significant co-expression clique of autism related genes, including Dpp6, Astn2 and Galnt13 (Menashe et al, 2013: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3723491/). A broader clique includes Dpp10, Nrxn1 and Rims3. 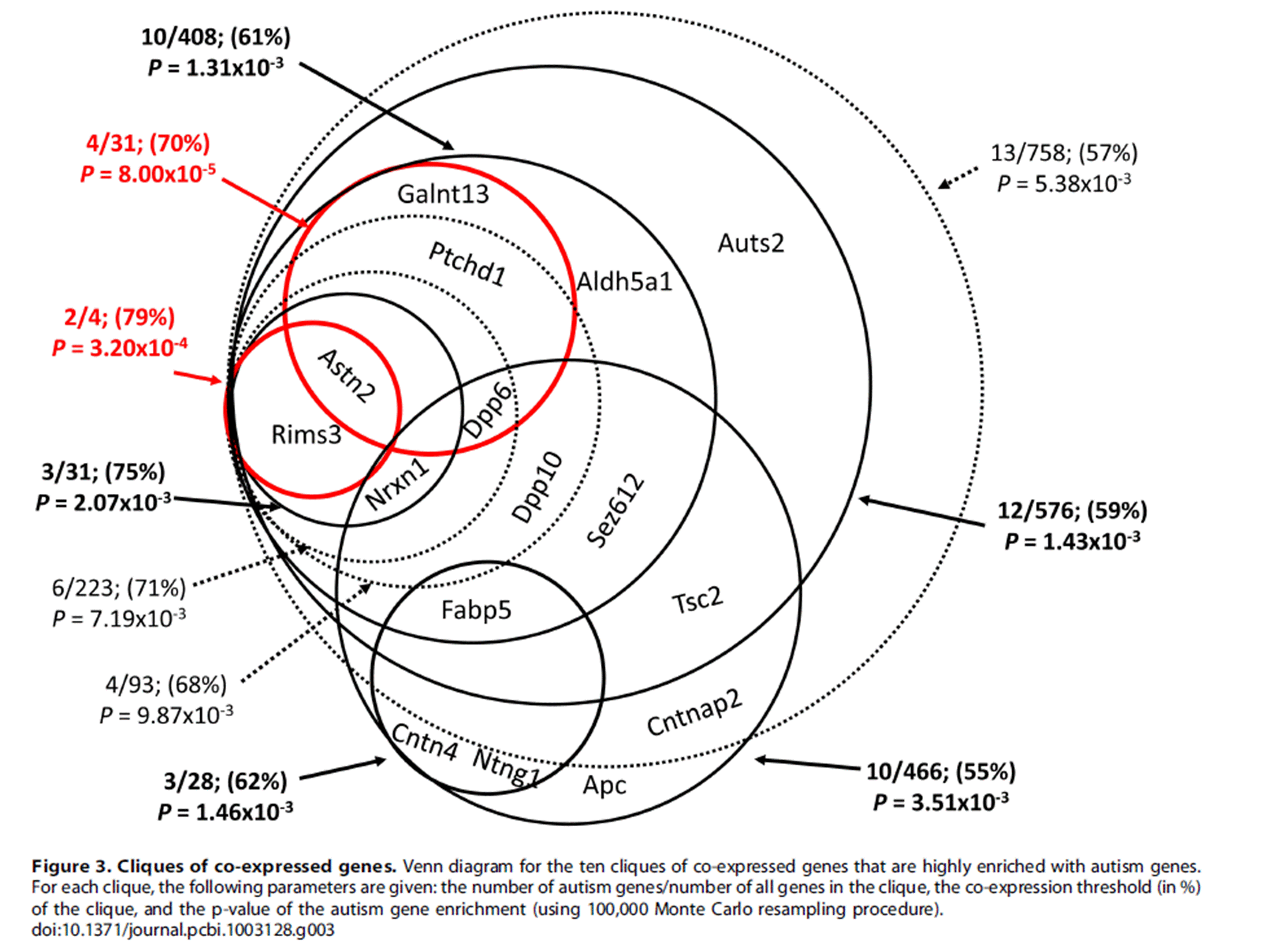
Expression of this clique is tightest within the cerebellum:

PTCHD1 as a target for FMR1 protein, FMRP?
A study for potential mRNA targets for FMRP, the mRNA-binding protein encoded by the Fragile X syndrome gene FMR1, suggests that a region in the 3’untranslated portion of the PTCHD1 mRNA binds to FMRP (coordinates ChrX:23414615-23414636; hg19)(Ascano et al, 2012: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3528815/).
PTCHD1 mRNA levels elevated by over-expression of MECP2 e1 isoform
In a study of the differential effects of over-expressing the two MECP2 isoforms, e1 and e2 (which differ chiefly in the N-terminal amino acid sequence, and are derived from alternative splicing of MECP2) in neuronal cell lines, using gene expression microarray analysis it was observed that there was a >5-fold up-regulation of PTCHD1 mRNA levels. However, this could not be validated by more accurate quantitative methods (qRT-PCR), as the expression of PTCHD1 was too low to accurately measure the change (Orlic-Milacic et al, 2014; http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3974668/). MECP2 is the gene responsible for the vast majority of cases of another intellectual disability disorder, Rett syndrome, and thus any connection between MECP2 and PTCHD1 at the molecular or cellular level would be of great interest.
PTCHD1 mRNA decreases in developing rat brain in response to NMDA antagonist ketamine
A study of the effects of ketamine on developing rat brain (post-natal day 7) showed a significant upregulation of NMDA receptor mRNA, and a downregulation of PTCHD1 mRNA (Shi et al, 2010: PMID:20080153; http://www.ncbi.nlm.nih.gov/pubmed/?term=20080153). Further studies would be required to validate the effect on PTCHD1, and to understand whether there is a direct interaction between ketamine and PTCHD1, or if this is a downstream effect of the upregulation of NMDA signalling activity, or another independent mechanism.
PTCHD1 and caveolin, and glucorticoid regulation.
In an MPhil research thesis submitted by Anna Mannion-Jones (supervisor ?) at the Manchester Centre for Nuclear Hormone Research in Disease (MCNHRD), University of Manchester, England, PTCHD1 was studied as a potential glucocorticoid regulated gene. The effects of dexamethasone (a potent glucocorticoid) treatment on PTCHD1 mRNA levels in caveolin-deficient cells were measured, and suggest that PTCHD1 levels increase, and are dependent on the presence of caveolin. See below for review.
PTCHD1 and Caveolin (by Bryan Degagne, M.Sc. student). Lipid rafts are specialized microdomains on the plasma membrane that are enriched in sphingolipids, cholesterols, and scaffolding proteins, and are believed to be involved in signal transduction processes (Simons & Toomre, 2000). One subtype of these microdomains, caveolae, are non-planar lipid rafts that appear as flask-shaped invaginations of the plasma membrane, and are uniquely rich in caveolin proteins (Fielding & Fielding, 2000). Caveolin proteins act as both a backbone for these structures and sites for other proteins to cluster around,thus providing an ordered lipid environment where related membrane-associatedproteins can aggregate and more readily interact with one another, making an ideal environment for signal transduction complexes to function efficiently (Brown & London, 2000). Given that caveolin proteins are also involved in trafficking proteins to and from the cell membrane (Stern & Marmelstein, 2010), they can act as powerful regulators ofmembrane-bound receptors and their related intra-cellular signalling complexes. Recently, PTCHD1 has been linked to Caveolin-1 (CAV1) based on differences in its expression in wild type and CAV1 knockout mice (Mannion-Jones, M.Phil. thesis, 2013), which supports evidence we have found through our own microarray experiments, though these data still need to be validated. As CAV1 seems to play a role in neuronal signalling and synaptic plasticity (Allen et al., 2007; Head et al., 2011; Liu et al., 2013), and has been linked to neurological abnormalities and disease (Trushina et al., 2006; Yeganeh et al., 2009; Stern & Marmelstein, 2010), it would be of particular interest to us if an interaction between PTCHD1 and CAV1 existed at the protein level. Though this has not yet been shown to exist, we can better understand the nature of any potential interactions by looking at proteins related to PTCHD1and their interactions with caveolins. For instance, Sonic hedgehog (SHh), the well-documented Hh ligand, associates with CAV1 in the Golgi apparatus to be transported to lipid raft domains at the cell membrane, where it can then be secreted into the extra-cellular matrix (Mao et al., 2009). Additionally, PTCH1, the known Hedgehog (Hh) receptor that has homology with PTCHD1, binds to CAV to be trafficked to and sequestered at caveolae lipid rafts. This interaction is also necessary for Smoothened to be transported to the membrane (Karpen et al., 2001). This suggests that CAV1 could be involved in the regulation of Hh signal transduction, and, as a likely Hh receptor itself, PTCHD1’s cellular localization may be similarly regulated by CAV1. However, unlike PTCH1, PTCHD1 includes a PDZ binding domain at its C-terminus, suggesting that it should interact with PDZ proteins, such as PSD-95. Interestingly, there is some evidence indicating that PSD-95 co-localizes with CAV1 based on its presence in buoyant caveolar fractions (Perez &Bredt, 1998; Bonds et al., 2010), and this seems to be regulated by palmitoylation of the N-terminal region (Craven et al., 1999), which is also necessary for PSD-95’s synaptic targeting (Ho et al., 2011; Zhu et al., 2013). If PTCHD1 does bind to PSD-95, this might represent a method for PTCHD1 membrane trafficking and regulation; however, the link between these proteins is still not known.
Ptchd1 knockout in mice
Early reports on the effects of knocking out the gene in mice, reported as a poster presentation at the Simons Center for the Social Brain by recent PhD graduate, Dr. Mike Wells from the Guoping Feng lab at Massachusetts Institute of Technology suggest that dysfunction of thalamic-cortical circuitry, and a defect in calcium homeostasis and burst firing in in the thalamic reticular nucleus (TRN) in particular. The mice showed cognitive impairment and motor defects, in addition to hyperaggression and attention deficit and hyperactivity.
http://web.mit.edu/scsb/posterSession.html?id=40
Abstract from grant application funded by Simons Foundation Autism Research Initiative, by Guoping Feng:
PTCHD4 is transcriptionally activated by p53, and suppresses Hh signalling (paper summarized by Bryan Degagne, M.Sc. student). (added 6 March 2015)
Chung, J. H., Larsen, A. R., Chen, E., & Bunz, F. (2014). A PTCH1 homolog transcriptionally activated by p53 suppresses hedgehog signaling. Journal of Biological Chemistry, 289(47), 33020-33031.PMID:25296753
Several Patched-related genes have been found in the human genome, though most are not well-known and their functions not entirely understood; however, the expression activity and potential function of one such relatively unknown Patched-related gene, PTCH53 (previously referred to as PTCHD4 due to its homology with PTCHD1), has recently been written about in a paper published in the Journal of Biological Chemistry. In the paper by Chung et al (2014), it was shown that the expression of PTCH53 is linked to that of the tumour suppressor gene TP53. The expression of PTCH53 markedly decreased in cells with a mutated TP53 gene when compared with unaffected cells and this was replicated in several human cell lines, though the differences in expression were most significant in cell lines derived from the central nervous system and skin. Additionally, administration of nutlin-3, a stimulator of p53-mediated transcription, resulted in a significant increase in PTCH53 and CDKN1A (another p53 target gene) expression.
On the other hand, the authors also linked PTCH53 expression to the Hedgehog signalling pathway, which was hypothesized based on its similarities to other Patched-related genes. It was shown that over-expression of PTCH53 has an inhibitory effect on GLI-mediated transcription similar to that of PTCH1, PTCH2, and PTCHD1 (shown previously by Noor et al., 2010), suggesting that PTCH53 could function as a Hedgehog receptor; conversely, transfection of PTCH53 inhibited GLI1, PTCH1, and PTCH2 expression, an effect that was also seen after nutlin-3 administration. P53 activity is known to suppress Hedgehog signalling, as evidenced by the effects of nutlin-3 administration; however, PTCH53 was not necessary for the suppression seen in this case, as blocking the expression of PTCH53 with shRNA did not alter the effects of nutlin-3 administration. Therefore, PTCH53 seems to be connected to both the Hedgehog and p53 signalling pathways, but it does not seem to be part of the mechanism by which p53 activity suppresses Hedgehog signalling. The authors suggested that these findings supported a role for PTCH53 in the DNA damage response or as a tumour suppressor, and potentially as a Hedgehog receptor in an auxiliary role distinct to certain physiological states.
Of particular interest to studies pertaining to PTCHD1, the authors also present independent replication of Gli suppression by PTCHD1, as previously described by our group (Noor et al, 2010).
